
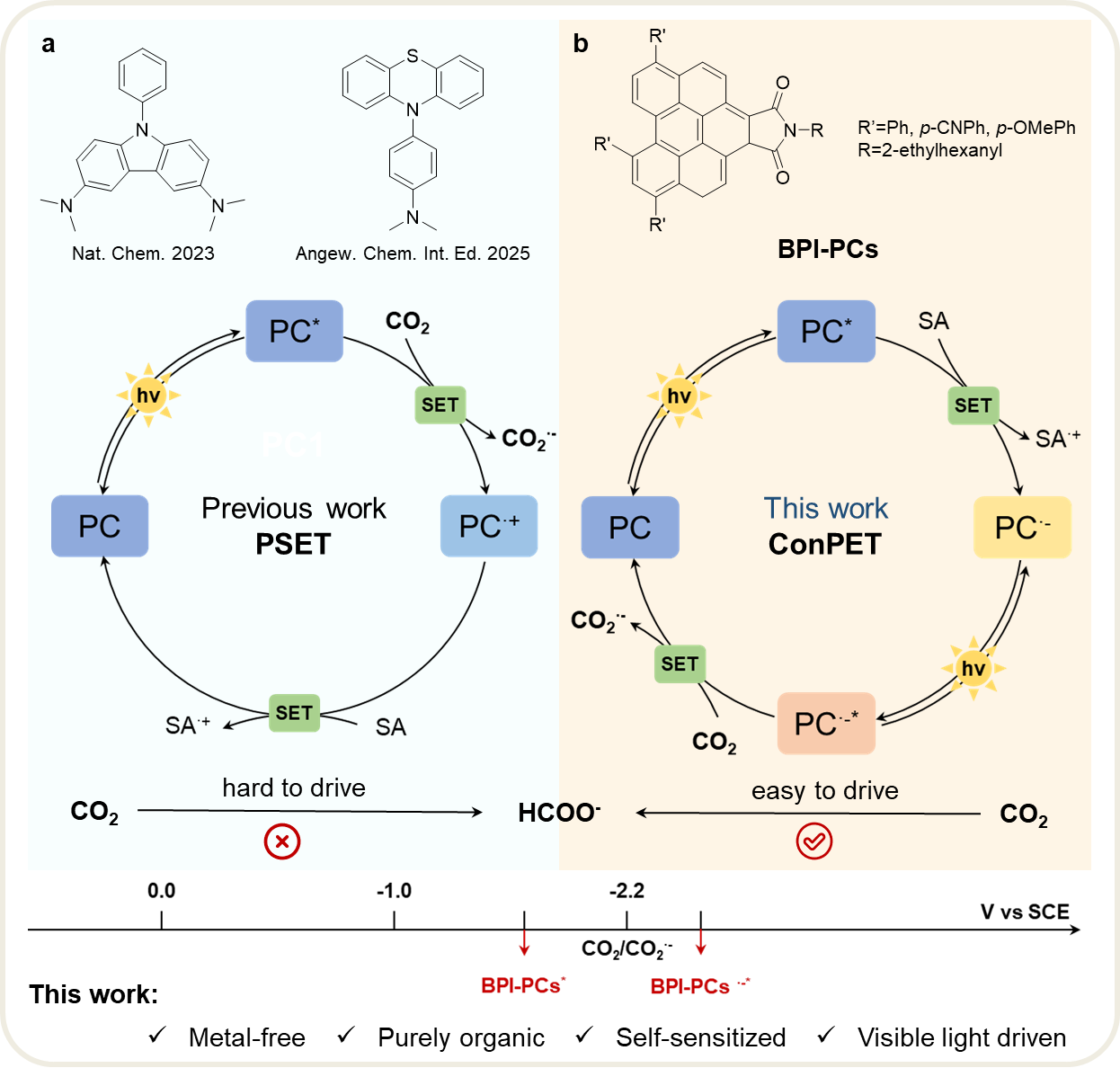
通过光催化二氧化碳还原反应(PCO2RR)将CO2转化为燃料和高附加值化学品,被认为是实现“碳中和”与太阳能化学储能的重要途径。然而,传统均相光催化体系通常需要独立的光敏剂(PS)和催化剂(Cat),体系复杂且容易发生电子回传等能量损失过程。相比之下,将光吸收、电子转移与催化活化功能集成于单一分子中的“自敏化分子光催化剂”,不仅能够简化催化体系,还可有效提高电荷利用效率。但目前该类体系仍然十分有限,尤其是可在可见光下运行的无金属体系更为罕见。受限于CO2活化所需的高还原势,许多无金属光催化体系只能依赖紫外光驱动,严重限制了太阳光利用效率。为突破这一能量瓶颈,连续光诱导电子转移(consecutive photoinduced electron transfer,ConPET)机制近年来受到广泛关注。在ConPET过程中,光催化剂能够在单个催化循环内连续吸收两个可见光光子,通过“多光子能量积累”获得超强还原能力,从而驱动传统单光子过程难以实现的高能反应。然而,将ConPET机制引入无金属、自敏化CO2光还原体系此前尚未见报道。
近日,彭慧晴课题组首次构建了基于ConPET机制的可见光驱动无金属自敏化CO2光还原体系。研究团队通过对一系列苯并苝酰亚胺(benzo[ghi]perylene imide,BPI)衍生物进行分子工程设计,成功发展出兼具光捕获、电子积累与CO2活化功能的单分子光催化剂,实现了可见光条件下CO2向甲酸盐(HCOO−)的高效高选择性转化。其中,含给电子甲氧基的BPI-OMe表现出最优催化性能,在可见光照射下甲酸盐生成速率达到27.51 mmol g−1 h−1,选择性超过99%,其表观量子效率(AQE)在405 nm下达到6.74%,并在多个可见光波段均保持较高活性。值得注意的是,该体系不仅能够在模拟太阳光下稳定运行,在自然太阳光照射条件下同样能够实现有效CO2转化,展现出良好的太阳能利用潜力。理论计算表明,给电子基团的引入显著提高了自由基阴离子态的电子密度,使其具备更低的激发能(2.98 eV)、更长寿命的电荷分离自由基阴离子态(τ = 806.94 ns)、更高能级的SOMO轨道(−3.47 eV)以及更强的CO2结合能力(−0.147 eV)。这些特征共同促进了电子积累与界面电子转移,使其更容易实现CO2活化。
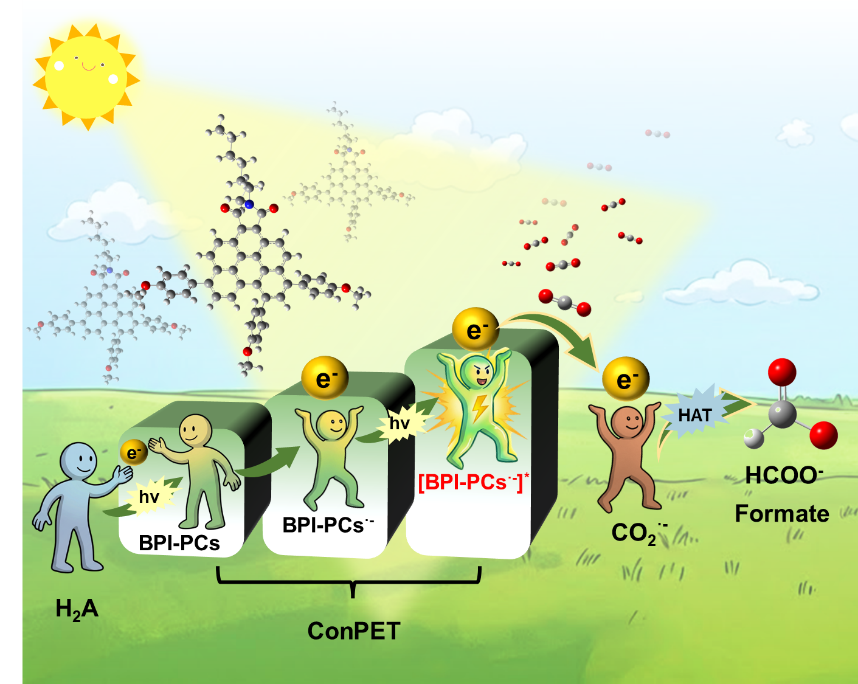
更重要的是,该工作系统揭示了ConPET驱动CO2还原的完整反应机制。荧光猝灭实验表明,抗坏血酸(H2A)能够向光激发态BPI-OMe发生电子转移,生成自由基阴离子BPI-OMe·−;随后,该自由基阴离子再次吸收光子,形成具有超强还原能力的激发态[BPI-OMe·−]*,进而将电子直接注入CO2生成CO2·−。原位ESR、ATR-FTIR以及超快瞬态吸收光谱进一步捕获并验证了CO2·−与BPI-OMe·−等关键中间体的形成过程。最终,CO2·−通过氢原子转移(HAT)过程生成甲酸盐,实现完整的双光子驱动CO2还原循环。该研究为无金属CO2光还原体系提供了全新的设计思路,展示了“多光子电荷积累”策略在高能化学转化中的巨大潜力。
该工作以题为“Metal-Free Photocatalytic CO2 Reduction to Formate Driven by Visible Light via a Consecutive Photoinduced Electron Transfer Mechanism”发表于Angew. Chem. Int. Ed.期刊,第一作者为北京化工大学软物质科学与工程高精尖创新中心硕士生殷贤君,通讯作者为北京化工大学化学学院刘宾副教授与软物质科学与工程高精尖创新中心彭慧晴教授。课题研究得到了国家重点研发计划与国家自然科学基金项目的资助。
文章信息:Xianjun Yin, Jingjie Hu, Heng Wu, Cui Xu, Bin Liu*, Hui-Qing Peng*. Metal-Free Photocatalytic CO2 Reduction to Formate Driven by Visible Light via a Consecutive Photoinduced Electron Transfer Mechanism. Angew. Chem. Int. Ed. 2026, e1083625.
原文链接:https://doi.org/10.1002/anie.1083625

